人们常说:“有啥别有病,没啥别没钱。”而对于一些罕见病患者来说,高昂的药费足以让他们的家庭步履维艰,甚至陷入绝望。
备受关注的2021年国家医保药品目录谈判中,曾经近70万元一支的诺西那生钠注射液,经过8轮“灵魂砍价”后,被“杀”到3.3万元,医保还可报销一定比例。
“这是我们晴晴(化名)的‘救命药’,也是给我们家最好的元宵节礼物!”2月15日,宁德师范学院附属宁德市医院儿科病房内,晴晴妈妈激动地说。
当天,6岁的晴晴在该院成功鞘内注射基因治疗药物“诺西那生钠”,成为“天价药”大幅降价并纳入医保目录后,宁德首位受益的患儿。

绝望:孩子患上罕见病
2016年,可爱的晴晴出生,给家里带来了无限欢乐。然而,在晴晴一岁多的时候,家人发现她走路姿势有些异常。
“刚开始以为是脚有问题,自己上网查了很多资料,带晴晴到医院进行了康复训练,但都没有效果。”晴晴的妈妈还带着晴晴去了福州的医院进行全面检查,也查不出病因,直到2017年1月,晴晴被确诊为脊髓性肌萎缩症。
据宁德师范学院附属宁德市医院儿科副主任医师钱丰介绍,脊髓性肌萎缩症(SMA)是一种罕见的染色体隐性遗传病。国内有2万至3万患者,根据现有资料,宁德目前有12名患儿。肌肉无力是这种罕见病最典型的症状。而目前唯一有效的药物,只有诺西那生钠注射液。
失望:无力承担天价药
“晴晴确诊后,诺西那生钠注射液在美国刚刚上市,一针要100多万元,我们根本负担不起。”晴晴的妈妈表示,他们一直在关注这个药。2019年4月,诺西那生钠注射液进入中国市场,但每针的价格仍然要近70万。
“如果一针能解决问题,我们砸锅卖铁也要给孩子用,但医生告诉我们,这个病的治疗是长期的。”晴晴妈妈说。原来,诺西那生钠注射液通过鞘内注射给药,可以改善患者运动功能、提高生存率,改变脊髓性肌萎缩症的自然病程,但注射第一针后,要在14天、28天、63天后再分别注射,以后每4个月注射一次,很多家庭确实无力承担。
晴晴的妈妈感觉,天快塌了。

希望:政策保障治疗路
“太好了,晴晴有救了!”2021年11月,在国家医保目录药品谈判现场,谈判代表通过8轮“灵魂砍价”,将诺西那生钠注射液的价格“杀”到3.3万元,晴晴的妈妈看着谈判视频激动地叫出了声。
紧接着,又传来好消息:2021年12月,诺西那生钠注射液进入国家医保名录。“从来没想到这个药能进医保,不管能报销多少,我们都感觉负担又减轻了好多。”晴晴的妈妈高兴不已,她立即打电话咨询哪几家医院有诺西那生钠注射液,“刚开始,省里只有少数几家医院有诺西那生钠注射液,为了治疗方便,我就打电话给市医院,问他们能不能采购这个药。”
在得知患者家属的需求后,宁德师范学院附属宁德市医院院长吴光辉高度重视,立即组织药事委员会和儿科医护团队开会,研究药物采购和治疗事宜,决定将诺西那生纳入该院药品采购目录。在吴光辉院长的牵头下,儿科主任郑明平立即开始组建治疗团队,药学部着手药品采购事宜。医院成立了SMA多学科治疗团队,包括神经发育专业组、儿童ICU专业组、影像学专业组、麻醉专业组、康复医学科专业组以及护理专业组。
2月15日9时45分,已接受局部麻醉的晴晴被医护人员抱着侧卧在床上,头膝卧位,儿科副主任医师钱丰留取和药物等量的脑脊液后,缓缓将5毫升诺西那生纳注射液注入晴晴腰椎蛛网膜下腔。10时,注射顺利完成,晴晴生命征平稳,无特殊不适。
“在晴晴之后,当天我们又为另一例脊髓性肌萎缩症患儿鞘内注射诺西那生钠。这两例治疗的成功开展也为今后更高难度的SMA鞘内注射治疗积累了宝贵的经验,铺筑了宁德市儿童SMA的治疗之路。”钱丰表示。
链接:
什么是脊髓性肌萎缩症(SMA)?
脊髓性肌萎缩症(Spinal Muscular Atrophy 简称:SMA)是一组会导致肌肉无力和萎缩的运动神经元病。运动神经起源于脊髓,控制着人体进行呼吸、爬、走、头颈控制以及吞咽等活动的肌肉。
SMA 对患者全身上下的肌肉都会造成侵害,患者下肢无力的情况通常较上肢更为严重。吞咽和呼吸功能(比如呼吸、咳嗽、清除气道分泌物)在中、重度患者中也会受到不同程度的影响,进而增加患者罹患肺炎和呼吸道感染的风险,也会造成患者睡眠时呼吸困难,需要进行呼吸支持。SMA 患者的认知功能、感官系统不受影响。通常,基于发病时间及所能达到的最大运动功能,患者被划分为 Ⅰ、Ⅱ、Ⅲ、Ⅳ四个类型。
SMA 的病因
SMA 是由于人体内被称作为“运动神经元存活 1 号”基因(SMN1)的缺失或异常(变异)所导致的。对于普通人而言,这一基因可以制造出一种被称为“运动神经元存活”(SMN)的蛋白。若此基因异常的话,人体内的 SMN 蛋白就会缺失或显著减少。
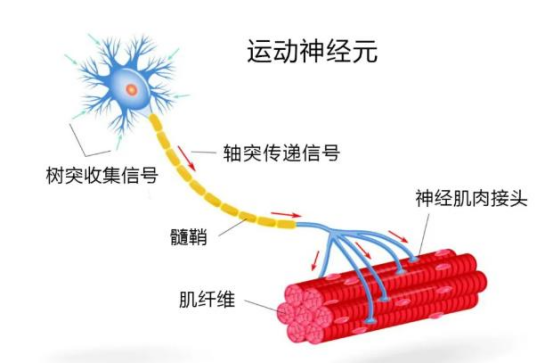
运动神经元是位于脊髓内的神经细胞,由它们所延伸出来的神经纤维通向身体各处的肌肉。SMN 蛋白对运动神经元的存活和健康至关重要,没有这种蛋白,神经细胞就有可能萎缩并最终死亡,进而导致肌肉的无力萎缩。随 SMA 患儿的生长,他们无力的肌肉很难满足日常活动的需要。这样的肌无力还会导致骨骼和脊柱的变形,进而加重呼吸方面的问题和进一步的身体功能丧失。
SMA 的遗传
最常见的SMA 是由 5 号染色体上的 SMN1(Survival Motor Neuron 运动神经元存活)基因缺失或变异所引起,因此也被称作 5q 型 SMA。
5q 型 SMA 属于常染色体隐性遗传病,即患儿父母双方都是致病基因携带者,孩子才会患病。但身为携带者的父母不会发病,也不会有任何 SMA 疾病症状。
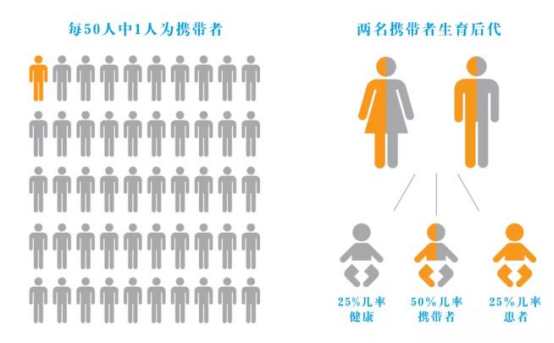
SMA 在新生儿中发病率为 1/6000-1/10000,常规人群中,约每40-50 人就有 1个是 SMA 致病基因携带者。父母同为携带者时,每次怀孕孩子都有:
• 25%的几率会是 SMA 患者
• 50%的几率会是 SMA 致病基因携带者(无症状)
• 25%的几率健康





