18岁花季姐姐、12岁少年弟弟,本该肆意奔跑、奔赴校园,却双双被困轮椅终身无法独行。姐弟二人先后确诊脊髓性肌萎缩症(SMA),全身肌肉进行性萎缩无力,肢体肌力持续衰退,无法自主站立行走,穿衣、进食、起居全部依赖家人全天候照料,大好人生被罕见遗传病牢牢禁锢。
最令人唏嘘的是,姐弟父母身体健康、无任何不适症状,无遗传病家族史,外表与常人毫无差别,却是隐匿的致病基因携带者。因为全家缺乏遗传筛查、产前诊断意识,两次生育接连中招,让两个孩子背负终身病痛,一个普通家庭就此陷入经济与精神双重困境。这场揪心的家庭悲剧,并非宿命天灾,而是完全可防、可控、可阻断的遗传遗憾。
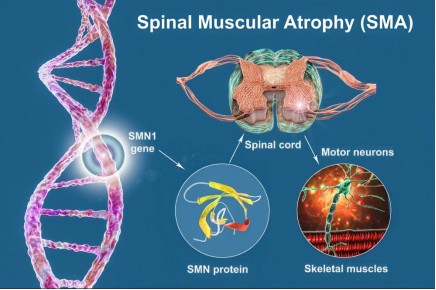
一、读懂SMA:藏在普通人身边的隐形遗传病
脊髓性肌萎缩症,简称SMA,是高发常染色体隐性遗传神经肌肉罕见病,致残风险极高。疾病根源为人体5号染色体SMN1基因缺失或突变,造成脊髓前角运动神经元变性坏死。
运动神经元是管控全身肌肉活动的核心单元,一旦受损,全身骨骼肌会持续萎缩、肌力不断下降,病情循序渐进加重,逐步剥夺人体行走、站立能力,晚期还会累及吞咽、自主呼吸功能,危及生命安全。
大众普遍误以为罕见病十分小众,但SMA离普通人极近:我国普通人群致病基因携带率达1/40-1/50,新生儿发病率约1/10000,是国内第二大常染色体隐性遗传病。临床将该病分为四型,其中青少年Ⅲ型隐匿性最强,患儿幼时发育完全正常,多在6-18岁缓慢发病,早期走路易摔跤、腿脚乏力,极易被误诊为缺钙、体质差,等到肌肉不可逆损伤,便彻底丧失行走能力,本次姐弟病例就属于该分型。
二、解惑误区:健康父母,为何生出患病孩子?
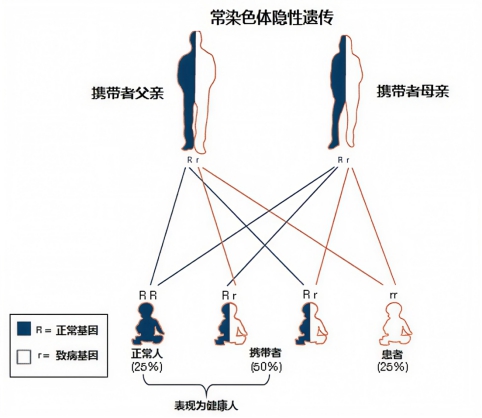
这是绝大多数患病家庭的共同疑惑,也是SMA最隐蔽的害人特性。SMA属于隐性遗传疾病,遗传规律固定清晰:
仅携带单条致病基因的人群,属于无症状携带者,身体无任何病症、体能不受影响,外观和健康人无异,绝大多数携带者终生不会知晓自身携带基因。
若父母双方均为无症状携带者,每一次自然生育,遗传概率恒定不变:25%概率生育患病子女、50%概率生育无症状携带者子女、25%概率生育完全健康子女。这户家庭父母同为隐性携带者,未做孕前筛查,两次生育均命中患病概率,最终酿成姐弟双双患病的悲剧。
三、临床现状:此病可控延缓,但无法根治
目前临床已有多款SMA靶向药物投入使用,药物可以延缓肌肉萎缩速度,延缓呼吸、吞咽功能恶化,降低重症并发症风险,但现阶段医学手段无法根治SMA。
针对姐弟这类已经发病多年、彻底丧失行走功能的青少年患者,药物仅能保命维稳,无法修复坏死神经元,更无法恢复行走能力。患病孩子终身无法自理,难以拥有正常学习、社交、人生,家庭需要长期承担靶向药、康复、护理高额开支,长期承受身心双重压力,负担难以承受。
四、优生两道防线:从根源阻断SMA发病
比起后期昂贵且有限的治疗,孕前筛查、产前诊断,是规避SMA患儿出生最高效、唯一彻底的方式。重点提醒:外表健康、无家族病史,不等于基因健康,超七成SMA患儿,都出生于健康普通家庭,备孕筛查不分人群。
第一道防线:孕前夫妻双方基因筛查
仅需无创抽血即可完成检测,便捷精准、无痛苦。单方携带致病基因,子代不会患病;双方同为携带者,即可提前规划生育方案,规避生育风险。
第二道防线:高危家庭孕期精准干预
夫妻双携带者受孕后,可在孕10-12周行绒毛穿刺、孕18-22周行羊水穿刺,判定胎儿患病情况;也可选择第三代试管婴儿技术,筛选健康胚胎移植,彻底阻断致病基因代代传递。
医生提醒,大多数遗传病都源于认知缺失和筛查缺位。建议所有SMA 患者、无症状携带者及高危备孕家庭,务必重视遗传咨询与优生筛查。携带者并非疾病患者,无需自卑焦虑,但一定要敬畏遗传规律,慎重对待生育问题。





